
What is Performance Evaluation and Why is it Important?
Under the In Vitro Diagnostic Medical Devices Regulation (EU) 2017/746 (IVDR), all In Vitro Diagnostic (IVD) devices must undergo a Performance Evaluation before they are placed on the market. This process is a continuous one, conducted throughout the device’s lifecycle, ensuring that it remains safe, accurate, and effective for its intended use. The Performance Evaluation assesses the Analytical Performance, Clinical Performance, and Scientific Validity, which together form the clinical evidence required by the IVDR.
This evaluation is vital for maintaining product safety and effectiveness and is a key part of ensuring that IVDs meet the necessary regulatory standards.
Steps Manufacturers Should Follow for Performance Evaluation Creation
Here are the 7 key steps involved in the Performance Evaluation process to help ensure that your IVD device complies with IVDR requirements:
Step 1: Assessing Data and Clinical Evidence
An important first step is determining the level of clinical evidence required to demonstrate IVDR conformity. To do this, you must assess your device’s intended purpose, device classification, state-of-the-art, and risks. This evaluation will guide you on the level of clinical evidence necessary to meet the General Safety and Performance Requirements (GSPRs) set out in Annex I of the IVDR. For legacy IVDs (those already marketed under the IVDD), a gap assessment should be conducted to identify any missing data. If gaps are found, you will need to generate the additional evidence required for compliance.
Step 2: Performance Evaluation Plan (PEP)
The Performance Evaluation process begins by establishing a Performance Evaluation Plan (PEP), which outlines how the evaluation will be conducted. The PEP will specify the key characteristics of the device, the methods used to demonstrate Analytical Performance, Clinical Performance, and Scientific Validity. The plan is guided by Annex XIII of the IVDR, which sets out requirements for Performance Evaluation, performance studies, and post-market follow-up.
Step 3: Scientific Validity
Scientific Validity refers to the relationship between the analyte (measured by the IVD) and a clinical condition or physiological state. To demonstrate scientific validity, you must prepare a Scientific Validity Report (SVR). This report should provide evidence that your device is state-of-the-art, explain its clinical benefit, and document the clinical background and significance of the analyte. A systematic literature search should also be conducted to support the device’s scientific validity.
If there are any gaps in scientific evidence, additional studies or expert opinions may be required.
Step 4: Analytical Performance
Analytical Performance involves demonstrating the IVD’s ability to correctly detect or measure a specific analyte. This is done through analytical performance studies that verify parameters such as:
- Analytical Sensitivity
- Analytical Specificity
- Limit of Detection (LOD)
- Limit of Quantification (LOQ)
- Precision and Accuracy
These studies should be detailed in an Analytical Performance Plan and summarised in an Analytical Performance Report (APR). If applicable, software characteristics for IVD Medical Device Software (MDSW) must also be considered, such as confidentiality and integrity.
These Analytical Performance parameters include the following and are set out in Annex I, section 9.1 a) of the IVDR:
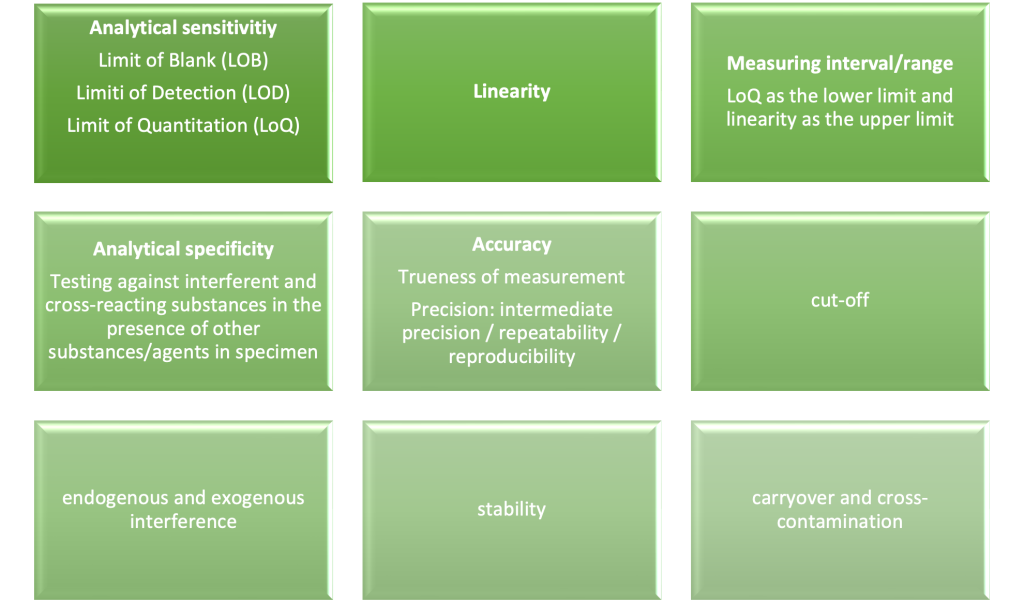
Step 5: Clinical Performance
Clinical Performance evaluates the IVD’s ability to accurately identify a disease or condition in the target population. This must be demonstrated using clinical performance studies, peer-reviewed literature, or published experience. The following clinical performance characteristics should be demonstrated:
- Diagnostic Sensitivity
- Diagnostic Specificity
- Positive Predictive Value (PPV)
- Negative Predictive Value (NPV)
Clinical performance must be documented in a Clinical Performance Study Plan and summarised in a Clinical Performance Report (CPR). These studies must adhere to the requirements outlined in Annex XIII Part A and Annex I, section 9.1(b) of the IVDR.
Step 6: Performance Evaluation Report (PER)
The Performance Evaluation Report (PER) consolidates all the data from the Scientific Validity, Analytical Performance, and Clinical Performance evaluations. This report ensures conformity with the relevant GSPRs, as set out in Annex I of the IVDR. The PER should also assess the device’s positive benefit-risk ratio in light of the current state of the art and medical technology.
The PER must be regularly updated as new evidence becomes available.
Step 7: Post-Market Surveillance (PMS) and Post-Market Performance Follow up (PMPF)
After your device has been released onto the market, ongoing Post-Market Surveillance (PMS) and Post-Market Performance Follow-up (PMPF) should be carried out. Regular review of PMS data helps determine if any updates to the PER are necessary. The PMPF plan outlines additional testing or monitoring requirements for the device’s safety and performance post-market.
Final Thoughts…
The Performance Evaluation is an ongoing process throughout the lifecycle of an IVD. It involves the continuous monitoring of safety, effectiveness, and performance through the collection of real-world data, feedback, and new scientific evidence. It is essential to keep the Performance Evaluation Plan (PEP) and Performance Evaluation Report (PER) updated to ensure the device remains compliant with regulatory standards.
Need Help Navigating the Performance Evaluation Process?
If you’re unsure where to start or need support in preparing the required documentation, LFH Regulatory can help. Our team of experts can guide you through the IVDR requirements, perform gap analyses, and provide consultancy services to ensure your IVD device gets to market smoothly.
Contact us today:
Phone: +44 1484 662575
Email: info@lfhregulatory.co.uk
Alternatively, simply complete the ‘Contact Us’ section below to find out how we can help you with your regulatory needs.
References:


