
Risk management is a core requirement of the EU Medical Device Regulation (MDR 2017/745) and In Vitro Diagnostic Regulation (IVDR 2017/746). It’s a central and continuous process that manufacturers must implement throughout the entire lifecycle of a medical device or in vitro diagnostic (IVD) device. Both MDR and IVDR refer to ISO 14971 as the gold standard for risk management. As a key component of the Product Life Cycle, risk management supports continuous safety and performance monitoring.
What is risk management for medical devices under MDR and IVDR?
Risk management is the systematic application of policies, procedures, and practices to identify, analyse, evaluate, control, and monitor risk associated with a medical device or IVD. Compliance with ISO 14971 is essential, ensuring risk management spans the entire lifecycle of the device – from design and development to manufacturing, post-market surveillance, and disposal.A well-defined and traceable risk management process is more than just a regulatory requirement, it’s a fundamental aspect of product development. By systematically identifying, assessing, controlling, and monitoring risks, it helps ensure device safety and effectiveness throughout its lifecycle.
The Management Team is responsible for:
• Defining the Risk Acceptability Criteria
• Ensuring Risk Management Planning is enforced
• Overseeing Benefit-Risk Analysis
• Reviewing Overall Residual Risk
What does MDR and IVDR require for risk management?
An effective risk management strategy is a cornerstone of regulatory success, reducing liability, enhancing safety, and streamlining market access. Under the MDR and IVDR, it is not just a requirement—it is a commitment to excellence in medical device innovation and patient care.
Applicable sections:
MDR: Annex I, Chapter I, Section 2-5
IVDR: Annex I, Chapter I, Section 2-5
Key obligations include:
• Establish and maintain a systematic risk management process.
• Conduct risk analysis to identify known and foreseeable hazards (including human factors risks).
• Estimate and evaluate the risks associated with these hazards.
• Implement risk control measures and verify their effectiveness.
These measures must:
• Reflect the state-of-the-art practices
• Remain effective throughout the device’s lifecycle.
• Minimise risks as much as possible.
• Determine if the residual risks are acceptable.
• Conduct benefit-risk analysis, ensuring benefits outweigh any remaining risks.
• Document all activities in a Risk Management File.
Definitions
| Term | Definition |
| Hazard | A potential source of harm |
| Hazardous Situation | A circumstance where people, property, or the environment is exposed to the hazard |
| Harm | Physical injury or damage to health |
| Risk | Combination of probability of harm occurring and the severity of harm |
| Risk Control | The process of making and implementing decisions to reduce the risk |
| Residual Risk | The remaining risk after applying the risk control measures |
| Benefit-Risk Analysis | Evaluating the medical benefits of a device against its residual risks |
A Risk Management File must contain the following elements:
• Risk management plan – Outlines the strategy and procedures for risk management activities.
• Hazard identification – Documentation of all known and foreseeable hazards.
• Risk analysis and evaluation – Assessment of identified risks, including probability and severity.
• Risk control measures and effectiveness – Description of implemented controls and verification of their impact.
• Residual risk evaluation – Analysis of remaining risks after mitigation.
• Benefit-risk analysis – Justification that benefits outweigh residual risks where applicable.
• Risk acceptability criteria – Defined thresholds for acceptable risk levels.
• Risk management report – Summary of the entire risk management process.
• Traceability to clinical and PMS data – Links to supporting clinical and post-market surveillance data.
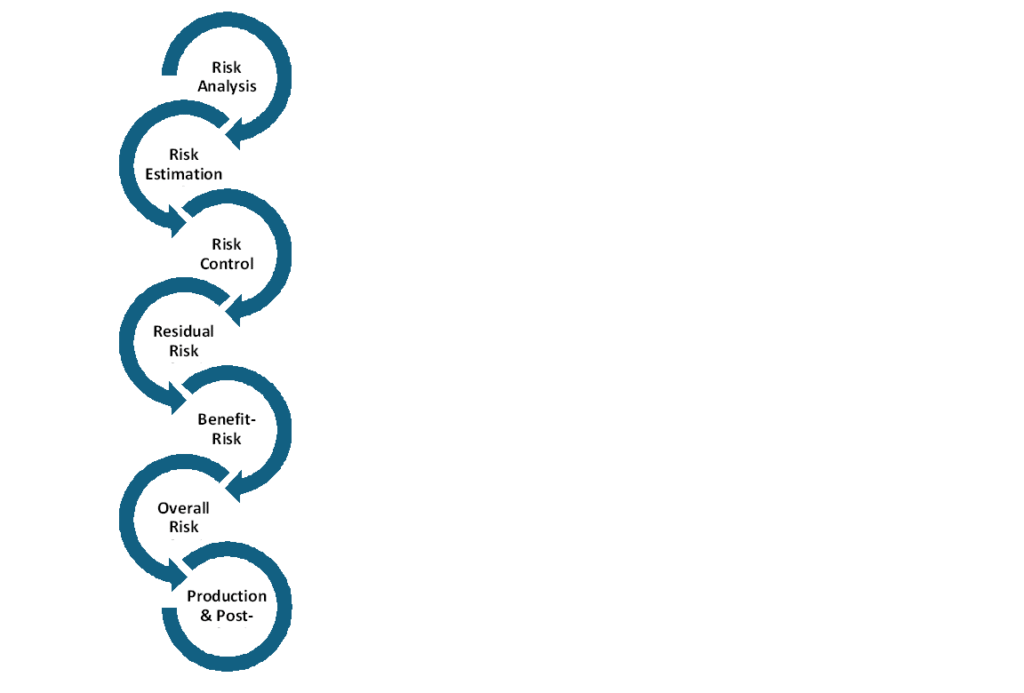
Risk Management Process Key Steps:
How does benefit-risk analysis work under MDR/IVDR?
The Benefit-Risk Analysis is defined under the MDR and IVDR as:
“The analysis of all assessments of benefit and risk of possible relevance for the use of the device for the intended purpose, when used as intended by the manufacturer.”
(Annex I, Chapter I, MDR and IVDR)
The Benefit-Risk Analysis involves a structured comparison of the device’s clinical benefits against its potential risks, ensuring that:
• The medical advantages for patients outweigh the identified risks under normal conditions of use.
• Compliance with General Safety and Performance Requirements (GSPRs) is demonstrated.
For the key considerations, when Residual Risk cannot be eliminated, manufacturers must provide clinical and technical justification that:
• The clinical benefits of the device outweigh the Residual Risk
• No safer alternative exists without compromising performance
This ensures patient safety while supporting innovation in medical technology.
How should risk be managed post-market under MDR/IVDR?
Risk management is a continuous process that extends well beyond product launch. The MDR/IVDR requires manufacturers to:
• Monitor Production & Post-Market Data – Systematically collect real-world performance information.
• Perform Trend Reporting – Analyse data to identify emerging risks or usage patterns.
• Update the Risk Management File – Ensure documentation reflects the latest safety assessments.
Once a device is commercialised, manufacturers must:
• Leverage PMS & Vigilance Data – Reassess risks based on clinical experience and incident reports.
• Address New or Evolving Risks – Take corrective action if the risk-benefit profile changes.
• Maintain Up-to-Date Documentation – Keep all risk management records current with new findings.
• This proactive approach ensures patient safety throughout the device’s entire lifecycle.
How does risk management support CERs and PERs?
The outputs of risk management play a critical role in the Clinical Evaluation Reports (CERs) under MDR and the Performance Evaluation Reports (PERs) under IVDR. These reports serve to
1) validate that clinical evidence adequately addresses all identified risks;
2) confirm the device’s safety and performance profile aligns with its risk-benefit assessment;
3) demonstrate risk control measures are supported by clinical data
The risk management process provides essential inputs that strengthen the clinical evidence base, ensuring comprehensive evaluation of device safety throughout its lifecycle.
Conclusion
Risk management under the MDR and IVDR is not simply a regulatory obligation—it’s a dynamic, continuous process that plays a critical role in ensuring both patient safety and device performance throughout the entire lifecycle. By proactively identifying, evaluating, and mitigating risks, manufacturers go beyond compliance to support innovation, reduce liability, and uphold trust in medical technology.
FAQ’s for Risk Management for MDR and IVDR Compliance
What is risk management for medical devices under MDR and IVDR?
Risk management is the systematic process of identifying, assessing, controlling, and monitoring risks linked to a medical device or IVD. It must cover the entire lifecycle, from design through post-market surveillance, and align with ISO 14971.
Why is ISO 14971 important for medical device risk management?
ISO 14971 is the internationally recognised standard for medical device risk management. It provides a structured framework that MDR and IVDR reference directly, ensuring manufacturers manage risks consistently and effectively.
What are the key requirements for risk management under MDR and IVDR?
Manufacturers must establish a risk management process, identify hazards, evaluate risks, implement and verify risk controls, assess residual risks, and conduct benefit-risk analyses. All activities must be documented in a Risk Management File.
What belongs in a Risk Management File?
A complete file includes a risk management plan, hazard identification, risk analysis and evaluation, details of risk control measures, residual risk evaluation, benefit-risk analysis, risk acceptability criteria, a risk management report, and traceability to clinical and post-market data.
How does benefit-risk analysis work under MDR/IVDR?
Benefit-risk analysis compares the clinical benefits of a device with its residual risks. If risks cannot be eliminated, manufacturers must show that the benefits outweigh them and that no safer alternative exists without compromising performance.
What happens to risk management after a device is launched?
Risk management continues post-market. Manufacturers must monitor real-world data, perform trend reporting, update the Risk Management File, and address new risks through corrective actions. This ensures patient safety throughout the device’s lifecycle.
How does risk management support Clinical Evaluation Reports (CERs) and Performance Evaluation Reports (PERs)?
Risk management outputs feed directly into CERs and PERs by validating that clinical evidence addresses all identified risks, confirming safety and performance profiles, and demonstrating that risk controls are supported by clinical data.
About the author – Dr Yupei Xiao

Dr Yupei Xiao specialises in regulatory and clinical affairs for in-vitro diagnostics (IVDs) and medical devices (MD), and she supports clients from startup stage to market submission. With expertise in performance evaluation, clinical evaluation, biological evaluation, and quality management systems, she helps companies navigate complex regulatory landscapes, ensuring compliance with MDR, IVDR, and ISO 13485:2016 standards.
With a Ph.D. in Respiratory Medicine and over a decade of experience in clinical trials, medical affairs and regulatory affairs, Yupei has supported startups and established firms in preparing technical files, conducting post-market surveillance, and implementing ISO-certified Quality Management Systems, led performance and clinical evaluations, supported international product registrations, and contributed to peer-reviewed publications and global regulatory submissions. Her work spans Medical devices, in-vitro diagnostics (IVDs) including the companion diagnostics, and Software as a Medical Device (SaMD), with a focus on EU and UK markets. Yupei combines deep technical knowledge with a practical approach to deliver compliant, timely outcomes. Her unique perspective bridges clinical science and regulatory strategy, making her a trusted advisor in the IVD and Medical Device industry.

