
Developing a medical device or an In Vitro Diagnostic (IVD) and bringing it to market can be a complex process filled with regulatory hurdles and quality management challenges. Understanding the main phases of medical device development and the regulatory requirements can streamline your efforts and ensure that your device meets all necessary standards for market approval. If you feel lost in the development process, you’re not alone. This blog will walk you through the critical stages of medical device development and the key documentation you need to comply with industry standards and regulations.
Understanding ISO 13485:2016 for Medical Device Development
ISO 13485:2016 is the globally recognised standard for Quality Management Systems (QMS) in the medical device industry. It ensures that medical devices meet regulatory requirements and satisfy customer needs. Implementing this QMS from the beginning of your product development is essential to streamline the process and avoid delays.
The QMS integrates procedures, work instructions, and controlled documents to guide and control all activities throughout the development of your device. The key to successful medical device development is ensuring that the right processes are implemented and followed early in the design phase.
Two essential processes that must be established early in the development of your device include:
- Design and Development: A robust design process that aligns with ISO 13485, Clause 7.3, and applicable regulatory requirements.
Risk Management: Align your risk management strategy with ISO 14971:2019 for medical devices and ISO/TR 24971 for IVDs.
What Are the Stages of The Design and Development Process?
The design and development process of medical devices typically follows a logical approach with several phases, as outlined in ISO 13485:2016. Below is an overview of these stages, which will guide you in navigating the process efficiently.
The Design Process in Figure 1 is a typical overview of the logical approach to apply to the phases of the design and development operation. In simple terms, the device requirements are developed and made to those requirements. It is then followed by verification and/or validation through to production and then manufacture.
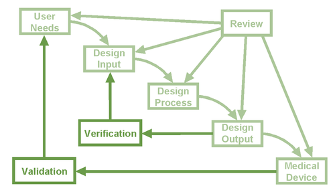
Figure 1: Design Process Overview
Design Reviews
Design reviews, also known as ‘stage-gate meetings’, are generally used to provide assurance that the activity or design phase has been satisfactorily completed and that the next stage of design can begin. They should be held at documented intervals throughout the design process, and the minutes recorded e.g., a review is conducted to ensure that design inputs are adequate and applicable to the device before work is carried out to convert them into design outputs.
Stage 1: Planning
A comprehensive Design and Development Plan is the foundation of the development process. This document outlines the scope, objectives, and deliverables of the project, and it must include:
- Project objectives and scope
- Design and development stages (inputs, outputs, verification & validation, design transfer)
- Responsibilities for the development team
- Regulatory requirements for the intended markets
- Risk management activities
- Supplier selection and management
Having a clear plan ensures that everyone involved in the process is aligned with the project’s goals, ensuring quality and timely delivery.
Stage 2: Design Inputs
Design inputs are the physical and performance requirements that define the functionality of your device. These inputs should be based on customer needs, user expectations, and regulatory requirements. They must also align with the intended use of the device.
Key design input elements include:
- User needs and expectations
- Regulatory and safety requirements
- Product performance specifications
- Usability standards
- Market-specific requirements for device classification
Stage 3: Design Outputs
Design outputs are the documented results of the design process, typically captured in a Design Traceability Matrix. Outputs include:
- Product specifications
- Raw materials, components, and sub-assemblies specifications
- Packaging and labelling specifications
- Design drawings, schematics, and manufacturing instructions
- Regulatory documentation for market submission
- Risk management reports and validation documentation (per ISO 14971)
These outputs demonstrate that the design and development process aligns with the initial requirements.
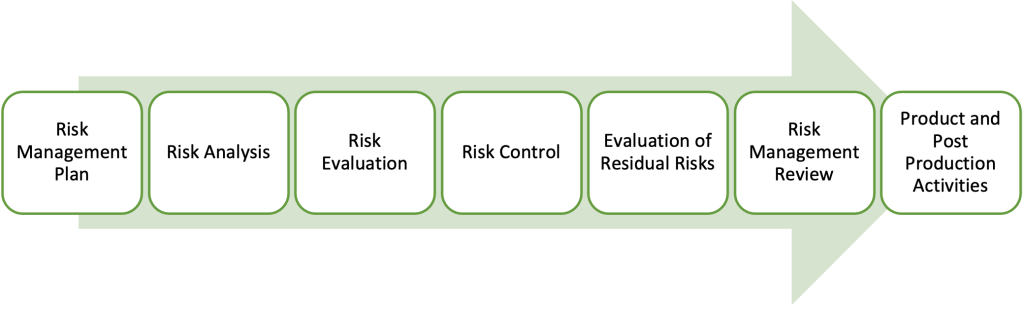
Figure 2: Risk Management Process Overview
Stage 4: Verification and Validation
Verification and validation are two crucial activities that ensure the device meets its defined input requirements.
- Verification confirms that the design meets the specified requirements.
- Validation ensures that the device meets the user needs and intended use.
Verification and validation documentation might include test protocols, clinical evaluation documentation, performance evaluation data, and clinical trials (if necessary).

Figure 3: Design Verification vs Design Validation
Verification and validation documentation can include, but is not limited to the following:
- Testing and analysis (Verification/Validation methods and acceptance criteria),
- Prototyping,
- Clinical Evaluation Documentation,
- Performance Evaluation Documentation,
- Clinical trials (where applicable).
Stage 5: Design Transfer
Once the device has passed all verification and validation tests, the design can be transferred to manufacturing. This stage includes:
- Final design approval
- Preparation of production records
- Manufacturing instructions
- Quality control plans
Design transfer documentation must be clear and comprehensive to facilitate the manufacturing process and ensure product consistency.
Stage 6: Control of Design Changes
Any design changes must be controlled through a formal change control process. Significant changes must be assessed for their impact on the device’s regulatory compliance, safety, and performance. These changes must be verified and validated before implementation.
Stage 7: Design and Development File
The Design and Development File (DHF) is a comprehensive record of all the design and development activities. It includes the documentation generated during each phase of development and provides evidence that all activities were conducted according to the QMS and regulatory requirements.
The DHF should include, but is not limited to:
- Test results (e.g., engineering, laboratory, simulated use)
- Biocompatibility and safety testing results
- Clinical evaluation reports
- Post-market surveillance plans
- Regulatory submission documentation
This file must be maintained and updated as the device progresses through development and its lifecycle.
Get Your Free Design Traceability Matrix
To help streamline your device development, we’ve created a free Design Traceability Matrix template. This tool will guide you in documenting your customer requirements, design inputs/outputs, and verification and validation activities.
Click here to send a request and we’ll email you a copy of the Design Traceability.
Conclusion: Ensuring Compliance and Success in Medical Device Development
Successfully navigating the design and development process of a medical device requires a clear understanding of the regulatory requirements and a well-structured approach to the design process. By following ISO 13485:2016 and ISO 14971:2019 standards, you can ensure that your device meets the necessary quality and safety requirements while preparing for a successful market launch.
At LFH Regulatory, we specialise in medical device regulatory compliance and can assist with designing and developing the required documentation. Contact us today to ensure your medical device meets regulatory expectations and enters the market smoothly.
Contact Us:
Phone: +44 1484 662575
Email: info@lfhregulatory.co.uk
References
- EN ISO 13485:2016+A11:2021 Medical devices – Quality management systems – Requirements for regulatory purposes.
- ISO 13485:2016 Medical devices. Advice from ISO/TC 210. A Practical Guide.
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices.
- Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices.
- ISO 14971:2019 Medical devices – Application of risk management to medical devices.
- ISO/TR 24971:2020 Medical devices – Guidance on the application of ISO 14971.
FAQs for Medical Device Development and ISO 13485 Compliance
What are the main stages of medical device development?
Medical device development typically includes planning, design inputs, design outputs, verification, validation, design transfer, control of changes, and maintaining a Design and Development File (DHF).
Why is ISO 13485:2016 important for medical device development?
ISO 13485:2016 sets the quality management framework that ensures medical devices meet safety, performance, and regulatory requirements. Implementing it early helps avoid delays and compliance issues.
What is the difference between design verification and validation in medical devices?
Verification checks that the design meets specified input requirements, while validation confirms the device fulfils user needs and intended use. Both are required for compliance.
What documents are included in a Design and Development File (DHF)?
A DHF contains records of the entire design process, such as test results, clinical evaluation reports, risk management documents, post-market surveillance plans, and regulatory submission files.
How does ISO 14971:2019 apply to medical device development?
ISO 14971:2019 defines how manufacturers should identify, assess, control, and monitor risks throughout the device lifecycle. It is a mandatory standard for regulatory compliance.
What role does risk management play in medical device design?
Risk management ensures hazards are identified early and mitigated effectively. It supports safety, regulatory compliance, and patient protection across design, production, and post-market stages.
What is design transfer in medical device development?
Design transfer is the stage where the approved design is handed over to manufacturing. It includes final design approvals, production records, manufacturing instructions, and quality control plans.

