
The AI Act & Medical Devices: What You Need to Know Now
Artificial Intelligence (AI) has been a hot topic in recent years, revolutionising industries worldwide. But as AI advances, so must the regulations that govern it. The AI Act (Regulation 2024/1689) introduces a structured legal framework to ensure AI-driven technologies, including medical devices, are safe, effective, and compliant.
Let’s break down what the AI Act means for medical device manufacturers, how it aligns with EU MDR (2017/745), and what you need to do to stay compliant.
What Is the AI Act?
The AI Act 2024/1689, approved in May 2024, is the first EU-wide regulation on artificial intelligence. It follows a risk-based approach, ensuring AI technology is trustworthy, safe, and transparent while fostering innovation.
Key highlights of the AI Act:
- Establishes a risk classification system for AI.
- Introduces mandatory requirements for high-risk AI systems.
- Encourages AI regulatory sandboxes for real-world testing.
- Applies to all AI systems operating in or supplying to the EU market (but not yet in the UK or US).
Does the AI Act Apply to Medical Devices?
Yes! Under Article 6, the AI Act classifies AI-driven medical devices as high-risk AI systems. That means medical device manufacturers need to comply with both EU MDR and the AI Act to obtain CE Marking and legally sell their products in Europe.
How AI Is Defined Under the AI Act
Under Article 3, AI is described as:
“A machine-based system designed to operate with varying levels of autonomy and that may exhibit adaptiveness after deployment… generating outputs such as predictions, content, recommendations, or decisions that can influence physical or virtual environments.”
If your Software as a Medical Device (SaMD) or AI-powered medical technology falls under this definition, you must comply with the AI Act.
Key Compliance Requirements: AI Act vs. EU MDR
The AI Act introduces new compliance requirements on top of existing EU MDR obligations. Here’s a quick comparison:
| Requirement | EU MDR 2017/745 | EU AI Act 2024/1689 |
| Quality Management System | ✔ | ✔ |
| Risk Management | ✔ | ✔ |
| Data Governance | X | ✔- NEW |
| Documentation Keeping | ✔ | ✔ |
| Automated generated logs | X | ✔- NEW |
| Technical Documentation | ✔ | ✔ |
| Cooperation with competent authorities | ✔ | ✔ |
| Post Market Surveillance | ✔ | ✔ |
| Corrective action and duty of information | ✔ | ✔ |
| EU Authorised representative | ✔ | ✔ |
| Conformity Assessment | ✔ | ✔ |
| Human Oversight | X | ✔- NEW |
| Transparency and provision of information to deployers | ✔ | ✔ |
| Accuracy, robustness and cybersecurity | ✔ | ✔ |
| Accessibility requirements | NEW | ✔- NEW |
| EU Declaration of conformity | ✔ | ✔ |
| EU AI Database registration | X | ✔- NEW |
| CE Mark | ✔ | ✔ |
| GSPRS | There is no GSPR requirement in the AI Act but the EU MDR GSPR list still needs to be followed. | |
What Does This Mean for You?
If you manufacture AI-powered medical devices, you must:
✅ Update your QMS to include AI-specific requirements.
✅ Implement data governance measures for AI models.
✅ Develop human oversight mechanisms to ensure AI systems remain under control. ✅ Ensure post-market monitoring includes AI-related risks.
Key Standards for AI Medical Device Compliance
To comply with the AI Act and EU MDR, manufacturers must consider the following standards:
✔ ISO 13485 – Quality Management for Medical Devices
✔ ISO 14971 – Risk Management for Medical Devices
✔ IEC 62304 – Medical Device Software Lifecycle Processes
✔ ISO/IEC 27001 & 27002 – Cybersecurity & Data Security
✔ ISO TR 34971 – AI-Specific Risk Management (Machine Learning)
✔ ISO/IEC JTC 1/SC 42 – AI Trustworthiness & Robustness
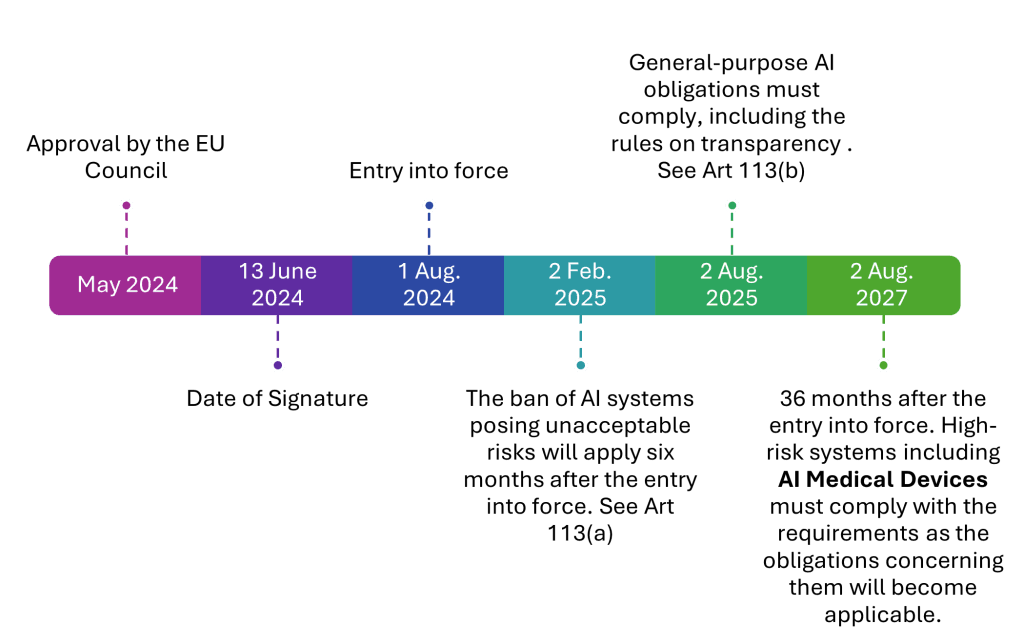
AI Act Compliance Timeline: Key Deadlines
2nd August 2027 – High-risk AI systems, including AI-powered medical devices, must be fully compliant with the AI Act.
What should you do now?
Assess your AI-powered medical devices to determine compliance needs.
Update your Technical Documentation to reflect AI-specific risks & controls.
Engage with Notified Bodies to ensure smooth CE Marking approval.
Here is a timeline explaining the journey from approval through to transition…
Challenges & Outstanding Questions
The AI Act is still evolving, and there are some unanswered questions:
- Is ISO 13485 alone sufficient for AI compliance, or will a separate AI Management System be required?
- How will substantial modifications to AI models impact compliance?
- Will medical device AI systems require predefined change control plans (PCCPs) similar to FDA requirements?
Keeping up-to-date with regulatory developments is crucial for staying compliant.
How LFH Regulatory Can Help
Navigating AI regulations can be complex, but LFH Regulatory is here to simplify the process:
✅ Regulatory Strategy Development – Tailored compliance plans.
✅ Technical Documentation & QMS Updates – Ensuring full alignment with EU MDR & AI Act.
✅ Notified Body Support – Helping you through the CE Marking process.
✅ AI Risk Management & Data Governance – Implementing AI-specific safeguards.
Stay ahead of AI regulations! Get in touch with our expert team today:
+44 1484 662575 | info@lfhregulatory.co.uk
FAQ’s for The AI Act 2024/1689
What is the AI Act 2024/1689?
The AI Act is the EU’s first regulation on artificial intelligence, approved in May 2024. It introduces a risk-based framework to ensure AI is safe, transparent, and trustworthy.
Does the AI Act apply to medical devices?
Yes. AI-powered medical devices and Software as a Medical Device (SaMD) are classed as high-risk AI systems under the Act. They must comply with both EU MDR 2017/745 and the AI Act to obtain CE Marking.
How does the AI Act define AI?
AI is defined as a machine-based system that can operate with varying autonomy, adapt after deployment, and generate outputs (such as predictions, decisions, or recommendations) that influence real or virtual environments.
What new requirements does the AI Act add for medical devices?
On top of MDR obligations, manufacturers must:
Implement AI-specific risk management and data governance
Ensure human oversight of AI systems
Keep automated logs of AI behaviour
Register in the EU AI Database
Meet accessibility and transparency requirements
When do medical devices need to be compliant with the AI Act?
By 2 August 2027, all high-risk AI systems, including medical devices, must comply.
What standards support AI Act compliance in MedTech?
Relevant standards include ISO 13485 (QMS), ISO 14971 (risk), IEC 62304 (software lifecycle), ISO/IEC 27001 (cybersecurity), and ISO/TR 34971 (AI risk).
How is the AI Act linked to CE Marking?
CE Marking remains essential under MDR. To gain CE approval for AI-powered devices, your QMS and technical documentation must now reflect AI-specific risks and controls.
What challenges remain unclear?
Questions include whether ISO 13485 will be enough for AI compliance, how major AI model updates will be handled, and if EU regulators will adopt predefined change control plans (PCCPs) similar to FDA requirements.
How can manufacturers prepare now?
Assess devices for AI risks and update documentation
Adapt QMS to include AI-specific controls
Engage with Notified Bodies early
Build AI risk management and oversight into your framework
Where can I get support with AI Act compliance?
Specialist consultants, such as LFH Regulatory, can help with strategy, QMS updates, risk management, and Notified Body engagement to ensure devices remain compliant.

Zara Malik
Zara works closely with a wide range of clients, supporting them and the wider team at LFH
in bringing medical devices and in vitro diagnostics (IVDs) to market. Her role spans internal
operations and project management, responding to a variety of client queries on quality and
regulatory matters, supporting the development of Quality Management Systems, Technical
Documentation, and assisting with Risk Management activities; ensuring compliance
throughout the product lifecycle.
With over 10 years of experience in the industry, Zara began her career in the laboratory of
an IVD company, where she quickly developed an interest in regulatory affairs. She went on
to specialise in risk management and internal auditing at a large medical device
organisation, before expanding her expertise into Technical Documentation and Post-Market
Surveillance during the implementation of the EU MDR and IVDR. Zara then joined a start-
up, gaining hands-on experience with Software as a Medical Device (SaMD) and AI/ML-
based medical technologies. She now brings this broad and evolving expertise to LFH
Regulatory, supporting clients across a range of complex and emerging regulatory
challenges.

